Yamamoto, T., Nagasaki, H., Yonemaru, J. et al. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genomics 11, 267 (2010). https://doi.org/10.1186/1471-2164-11-267
作者首先利用全基因组测序,测定了日本水稻品种Koshihikari。随后,将其与已发表的Nipponbare序列进行比对,确定了67051个SNP位点。随后,利用1917个SNP位点,对151个Japanese cultivars进行芯片(high-throughput typing array)测定。利用这些数据,作者确定了从traditional landraces中继承而来的、60.9%的Koshihikari基因组、18个共有单倍型块的祖源,并预测现代育种实践普遍减少了遗传多样性。
作为结论,作者评估了相关水稻品种的遗传组成,并借助系谱信息阐明了育种中的染色体重组的动力学过程。作者还发现了及个基因组区域的遗传多样性降低,并猜测时育种中的选择造成的。最后,作者认为,基于SNP的系谱单倍型的确定,将促进水稻和其他作物的育种。
技术细节1. 测序与比对
统计了:
- 测序深度分布与平均值
- contig的数量、长度分布
- About 76.0 Mb of the Nipponbare genome was not covered by the short Koshihikari reads.并进行了reads的分类与占比
- 无法比对到参考基因组上的读段统计
对于成功比对的部分,统计了各染色体SNP密度分布,并从全部SNP中随机选取了64各,用traditional Sanger method using a capillary sequencer进行验证,已验证找到的SNP的可靠性。下图为SNP密度分布的部分截图。
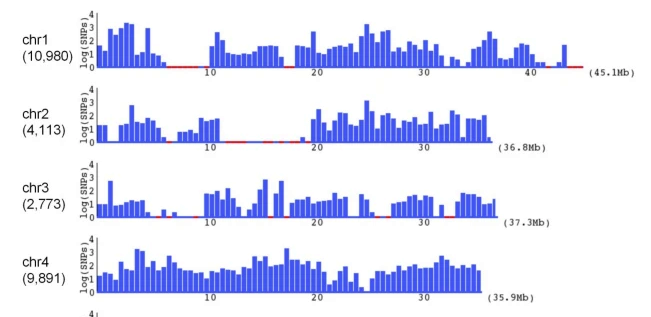
模拟了测序深度、基因组覆盖度和检测到的SNP数量三者之间的关系,得出10x的测序深度足够了。
统计了SNP的突变类型与占比。
在芯片测序部分,作者在芯片设计上,最初设计了2688个SNP,并在筛选后保留下了1917个
2. 系谱单倍型确定
2.1 结论性部分
选择了日本150年中151个代表性cultivars进行芯片测序。提出由于这些品种是育种系(breeding lines),因此不适用种群结构分析等方法。下图为STRUCTURE软件分析的结果。
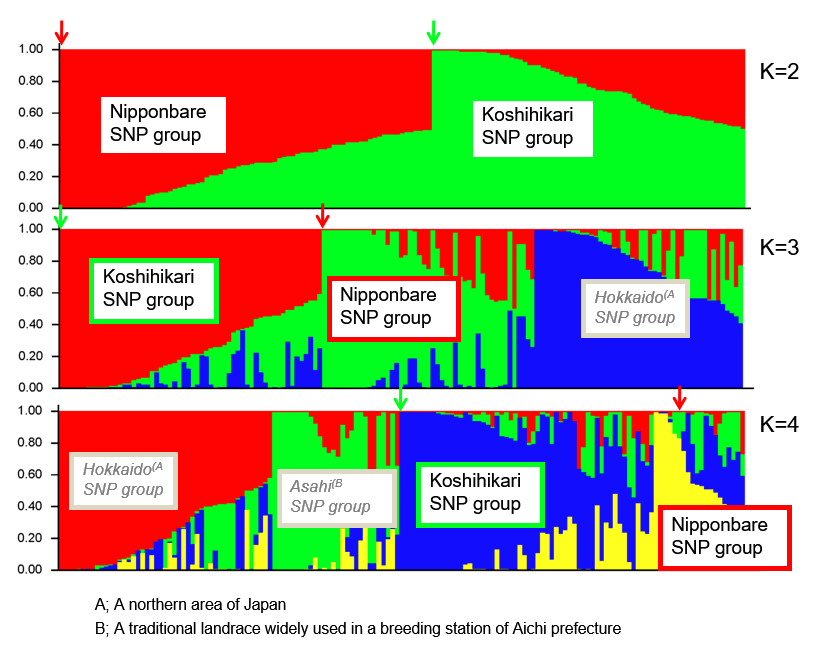
作者根据育成年份将这些品种大致分为三类。其中,第二组的育种时期,高产主要目标;第三组所在时期的目标则开始演变为品质。
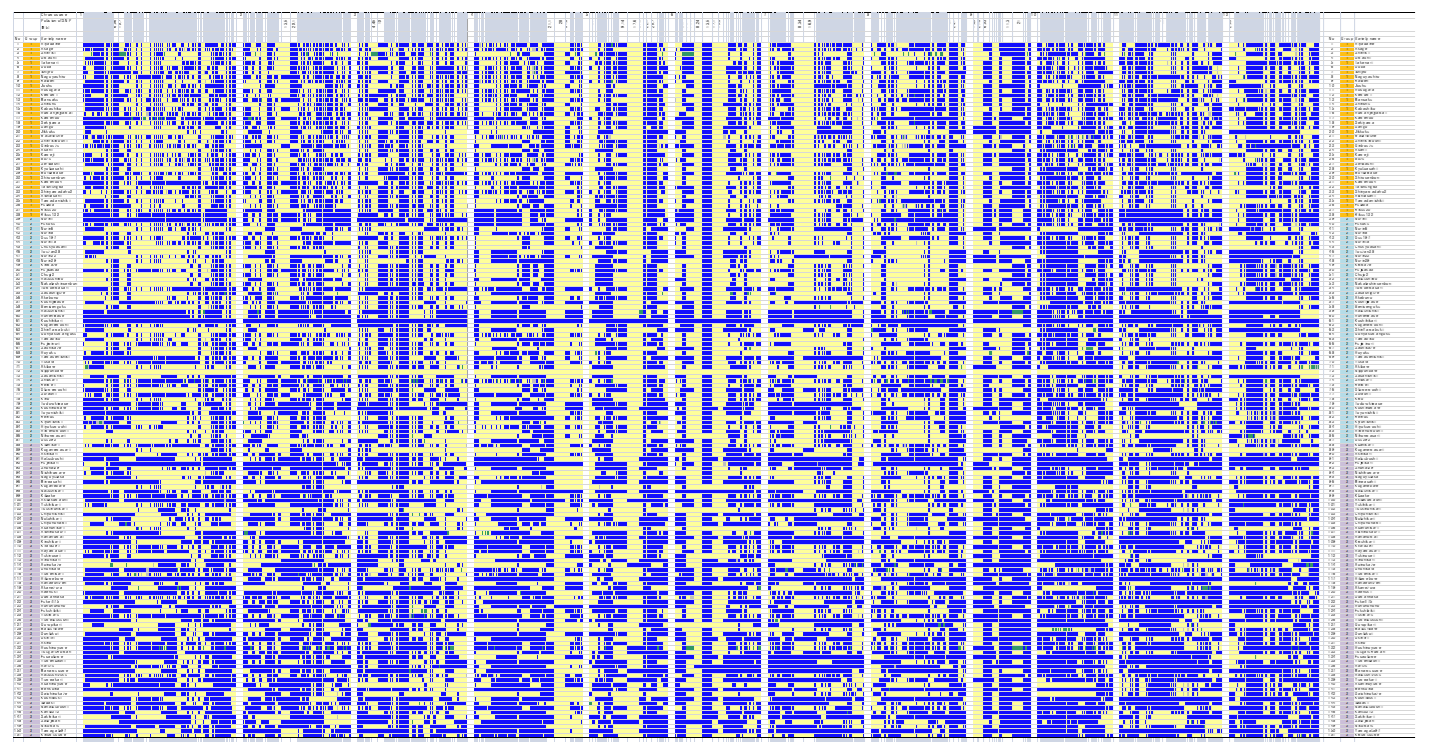
根据等位基因的对比,作者根据等位基因基因型的对比,确定了各品种的基因组组成(下图)。
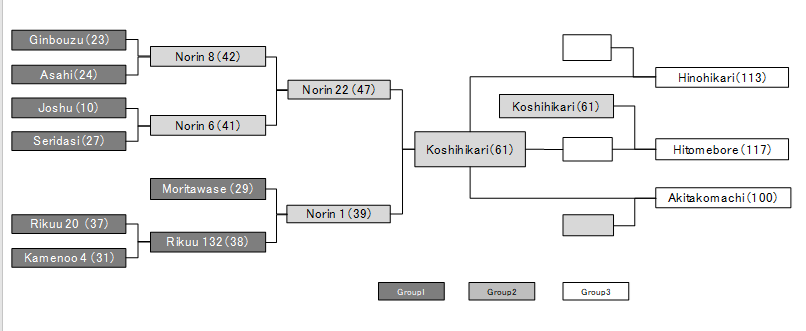
下图展示了Koshihikari前后几代的系谱关系
下图展示了基于SNP和系谱信息估计出的Koshihikari基因组组成。尽管系谱单倍型的可靠性受到单倍型块内SNP数量的影响,作者仍确定了60.9%的Koshihikari基因组的系谱单倍型祖源,找出了6个landraces祖先(下图A)。
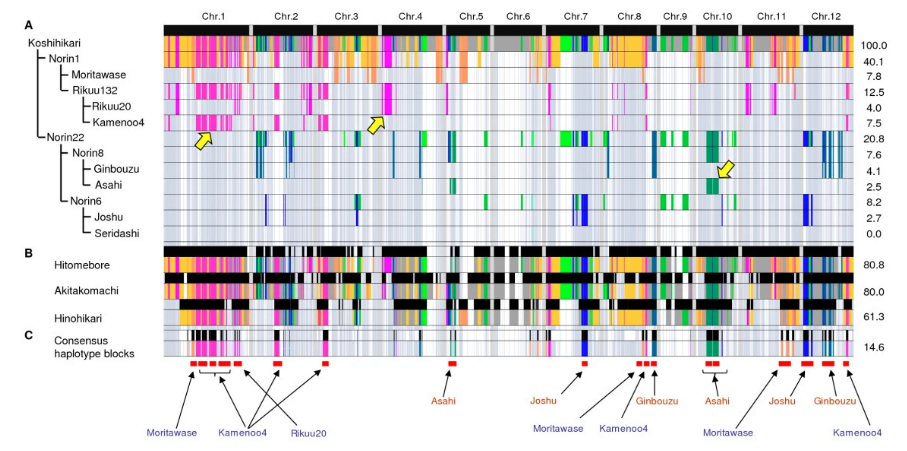
随后,作者找了几种通过与Koshihikari杂交而育成的、广泛种植的日本水稻品种,并评估了他们从Koshihikari中继承来的基因组片段的位置与比例(上图B)。上图C是一些统计信息。
2.2 技术细节
作者提到,有文章(doi: 10.1101/gr.089516.108)提出了一种基于重组自交系全基因组重测序的高通量的基因分型和重组点估计的方法。但“该方法只能用于基因组序列已被解码的品种,这使得很难将其方法应用于大量独立系或品种上”。因此,作者首先进行了全基因组SNP检测(Koshihikari),然后进行了芯片测序(151个品种)。芯片测序的位点是根据WGS测序找到的。
单倍型块确认上,作者对比了Koshihikari和其祖先的基因型,当相同的基因型块超过500kb时,认为是一个单倍型块。
3. 现代水稻育种与基因组多样性变化
作者通过滑动窗口,估计每单位基因组区域所具有的单倍型数量,并将其作为基因组多样性的指标。
窗口大小由LD估计。We calculated that the LD decay was 2 Mb